허가 의약품 품목 취하·취소 시 추가 동의서 이용해 신청 불가
저함량 품목도 고함량 품목과 동일하게 자료 사용 동의로 판단
[메디칼업저버 정윤식 기자] 시행 시기부터 방법까지 무수한 추측이 많았던 공동생동 1+3 제한법이 공포 후 즉시 시행으로 확정됐다.
식품의약품안전처는 지난 20일 동일한 생물학적 동등성 시험자료를 이용한 의약품 품목 수 1+3 제한을 담은 약사법 개정안을 공포했다.
이번 개정안으로 인해 허가 시 동일한 제조소, 제조방법 등을 가진 의약품에 대해서는 3회에 한해 생물학적 동등성 시험자료의 추가 이용이 가능하다.
종전에는 생물학적 동등성 시험자료의 공동 이용과 사용 횟수 제한 등에 대한 별도의 근거가 없었다.
결국 제약사들이 가장 주목했던 시행 시기의 경우 공포 즉시로 결정됐고, 이 외에 △공조대조약 고함량 품목과 저함량 품목의 차이 △제조소 변경허가 신청에 따른 적용 대상 △공동개발 중인 의약품의 해당 여부 등 주요 쟁점에 대한 식약처의 해석을 분석했다.
허가 의약품 품목 취소 시 추가 동의서 이용 불가
우선, 이번 개정 규정 1+3 시행 전에 특정 임상시험자료를 사용하도록 여러 번 동의한 경우라도 시행 후부터 3회에 한해 해당 임상시험자료를 사용할 수 있도록 추가 동의가 가능하다.
즉, 개정 전에 자료이용 5회에 동의했더라도 개정 후 3회를 추가하면 총 8회가 되는 것이다.
단, 개정 규정 시행 이후 품목허가를 신청하거나 품목신고를 한 경우부터 적용하도록 규정하고 있으므로 시행 전에 이미 품목허가를 신청해 허가 심사 중인 의약품에 대해서는 적용하지 않는다.
주의할 부분은 임상시험자료 사용을 동의 받아 허가된 의약품 중 일부품목이 취하·취소된 경우라도 해당 임상시험자료를 이미 작성했던 자는 추가로 해당 자료의 사용에 동의할 수 없다는 점이다.
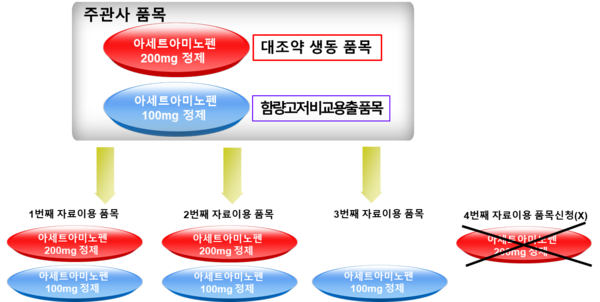
저함량 품목의 경우 고함량 품목의 생물학적 동등성을 기반으로 허가받은 것이기 때문에 저함량, 고함량 품목은 묶음으로 자료 사용이 동의됐다고 본다.
다시 말해 저함량이든 고함량이든 3회의 생물학적 동등성시험자료 사용의 동의가 있으면 추가로 자료사용 동의가 가능하지 않다는 것으로, 함량 차이가 '3회'라는 횟수에 영향을 미치지 않는다는 의미다.
제조소를 변경해 변경허가를 신청할 시, 변경된 제조소에서 생산된 제품의 임상시험자료를 기반으로 해 변경허가를 받게 되므로 변경된 제조소의 임상시험자료를 작성한 자의 자료사용 동의에 따라 변경허가가 가능하게끔 했다.
이 경우에도 임상시험자료를 작성한 자는 개정 규정 1+3 시행 후부터 3회에 한해 해당 임상시험자료를 사용할 수 있도록 추가 동의가 가능하다.
생물학적 동등성시험자료의 범위에 '생체를 이용하지 아니한 시험자료로서 생물학적 동등성시험 자료를 갈음해 제출 가능한 자료'를 포함해 규정하고 있기 때문에 생물학적 동등성시험자료의 제출 대신 비교용출·비교붕해·이화학적동등성 시험자료 등을 제출하는 경우에도 개정 규정이 적용된다.
동일 임상시험자료를 이용한 품목 수를 제한하는 규정은 원칙적으로 전문의약품 중 '첨단재생바이오법'에 따른 첨단바이오의약품, 생물학적 제제를 제외한 의약품에 대해서 모두 적용된다.
공동개발의 경우 임상시험 계획 승인 여부가 중요
가장 관심이 많았던 부칙 중 하나인 공동개발과 관련해서는 임상시험계획 승인이 가장 중요한 조건이다.
먼저 1+3 시행 당시에 다수의 의약품 제조업자가 공동개발하기로 하고 생물학적동등성시험을 포함해 임상시험 계획 승인을 받은 의약품인 경우, 공동개발한 의약품 제조업자가 공동개발 사실을 신고하면 이번 개정 규정을 적용 받지 않는다.
하지만 개정 규정 시행 전부터 공동개발 중이었다고 해도 시행 당시에 임상시험계획이 승인되지 않았다면 반드시 규정된 내용을 적용 받게 된다.
아울러 규정 시행 전에 공동개발하기로 하고, 임상시험계획을 승인 받은 의약품에 대해서는 공동개발하기로 한 '의약품 제조업자'에 한해 해당 규정을 적용받지 않도록 규정하고 있지만, 규정의 취지상 의약품 제조업자 외에 '위탁제조판매업자'도 이에 포함해 해석할 수 있을 것으로 판단된다.
시행 당시 의약품을 공동개발하기로 하고 임상시험계획 승인을 받은 자가 해당 사실을 신고하려는 경우, 공동개발 사실이 명확히 기재된 계약서 등을 공증 받아 제출해야 한다.
이 때 공동개발 계약서에는 공동개발하려는 제품을 특정할 수 있는 정보 및 공동개발 추진 시 업무 또는 비용의 분담에 관한 사항이 포함돼야 하며, 계약서상의 공동개발자 정보는 이후에 품목허가를 신청하는 의약품의 제조업자 정보와 동일해야 한다.
공동개발 사실을 신고하려는 경우에는 의약품안전나라 홈페이지에서 신고 내용을 작성한 후 관련 자료를 첨부해 제출하면 된다.
이번 개정은 임상시험자료를 작성한 자 즉, 임상시험계획을 승인받은 자가 3회에 한정해 타인에게 해당 자료의 사용에 동의할 수 있도록 한 것이다.
이에 임상시험자료를 작성한 자가 아닌 공동개발자는 다른 업체에게 해당 임상시험자료를 사용하도록 동의할 수 없다.
한편, 이번 약사법 개정안에는 1+3 외에 △중앙임상시험심사위원회 근거 마련 △백신안전기술지원센터 설립근거 마련 △거짓·부정한 허가 및 국가출하승인에 대한 제재 강화 등의 내용이 포함됐다.